Yu Du1, Fakang Xie1, Mengfei Lu1,2, Rongxian Lv3, Wangxi Liu1,2, Yuandong Yan1, Shicheng Yan1*, and Zhigang Zou1,2
1Collaborative Innovation Center of Advanced Microstructures, National Laboratory of Solid State Microstructures, Eco-materials and Renewable Energy Research Center (ERERC), College of Engineering and Applied Sciences, Nanjing University, No. 22 Hankou Road, Nanjing, Jiangsu 210093, P. R. China.
2Jiangsu Key Laboratory for Nano Technology, Nanjing University, No. 22 Hankou Road, Nanjing, Jiangsu 210093, P. R. China.
3Industrial Center, Nanjing Institute of Technology, No. 1 Hongjing Avenue, Nanjing, Jiangsu 211167, P. R. China.
*Correspondence: yscfei@nju.edu.cn
Abstract: Compressive strain, downshifting the d-band center of transition metal oxides, is an effectiveway to accelerate the sluggish kinetics of oxygen evolution reaction (OER) for water electrolysis. Here, we find that anisotropic thermal expansion can produce compressive strains of the IrO6 octahedron in Sr2IrO4 catalyst, thus downshifting its d-band center. Different from the previous strategies to create constant strains in the crystals, the thermal-triggered compressive strains can be real-timely tuned by varying temperature. As a result of the thermal strain accelerating OER kinetics, the Sr2IrO4 exhibits the nonlinear lnjo- T−1 (jo, exchange current density; T, absolute temperature) Arrhenius relationship, resulting from the thermally induced low-barrier electron transfer in the presence of thermal compressive strains. Our results verify that the thermal field can be utilized to manipulate the electronic states of Sr2IrO4 via thermal compressive strains downshifting the d-band center, significantly accelerating the OER kinetics, beyond the traditional thermal diffusion effects.
Introduction
Electrochemical water splitting driven by renewable electricity provides an ideal strategy for sustainable production of clean hydrogen fuel from water. However, the high overpotentials for oxygen evolution reaction (OER) in the water electrolysis pose a bottleneck for large-scale hydrogen production. Therefore, it is urgent to develop efficient and stable OER catalysts to overcome this challenge. Electronic states of materials, determining the energetics during the reagent adsorption, the evolution of intermediates, and the product desorption, are directly related to the OER kinetics. Strain engineering is especially effective in optimizing the d-band center of transition metal-based catalysts via compressive or tensile strain in crystals. Generally, tensile strain reduces the overlap of the wavefunctions and thus gives rise to the narrowing of the metal d band and an upshift of d-band center.In contrast, compressivestrainhas the opposite effect, causing a broadening of the d band and a downshift of the d-band center. For OER, the d-band center is closely related to the interaction energy betweenadsorbate states and the metal d states, thus determining the OER kinetics. Conventionally, various straining methods, such as lattice mismatch,doping heteroatoms,morphology controlling, and introducing defects, were proposed to produce the constant strains in the crystals. In these methods, the constant strain is mainly determined by the compositions, microstructures and synthesis conditions of materials. Additionally, the variable strains can be in situ generated during the service of materials, in which the materials directly grow or are coated onto the flexible substrates to form a flexible film which can be subjected to tensile or compressive loading from the external forces. Obviously, although the variable strain is beneficial to optimize the electronic states, the dependence of complex preparation processes and state-of-the-art equipment limits its practical applications.
In order to promote the applications of strain engineering in OER, there is an urgent need to find a simple method to generate variable strains in materials. Heat is a common source of energy that can be easily utilized and modulated to produce various thermophysical effects, such as thermal expansion, thermal diffusion, and thermal phase transition, which are useful in energy conversion fields. In particular, it was demonstrated that the anisotropic thermal expansion typically originates from the thermal-induced elementary microstructural deformation caused by transverse vibrations of the bridging atoms, probably producing strains. Accordingly, it is possible to utilize the thermal expansion effects as a convenient route to generate variable strains in OER catalysts. Here, we heat the Sr2IrO4, an anisotropic thermal expansion material, to generate thermal strains for verifying the effectiveness of this idea in regulating electronic states of materials. Under thermal stimulation, thecompressive strains of the IrO6octahedra in Sr2IrO4 catalyst can be created, consequently downshifting the d-band center. As a result, the binding strength between OER intermediates and the Ir active species was optimized to meet the low-barrier catalytic reaction described by Sabatier principle, thus accelerating OER kinetics following the nonlinear Arrhenius relationship. Our findings prove that thermal stimulation producing compressive strains is an effective route to tune the electronic states of Sr2IrO4 with anisotropic thermal expansion, significantly increasingOER performances, beyond the traditional thermal diffusion effects.
Results
Thermal strains in Sr2IrO4
In order to confirm the structures and compositions of Sr2IrO4 during temperature-dependent OER, we carried out an OER i-t test at 1.45 V at90 oC for 1 h. Afterwards, the X-ray diffraction (XRD) and scanning electron microscope (SEM) analyses indicated that the electrode is electrochemically stable Sr2IrO4 particles with tetragonal structure (JCPDS Card No. 09-0099) and particle size of 200 - 300 nm (Supplementary Fig. 1a,b). The high-resolution transmission electron microscope (HRTEM) lattice image confirmed that the Sr2IrO4 particle is highly crystalline and exhibits the continuous lattice fringes of 0.32 nm for (004) facet extending through the whole particle, evidencing that the particle with irregular shape is an undeveloped single crystal, as confirmed by the fast Fourier transform image (Fig. 1a). To disclose the growth mechanism of the Sr2IrO4 single crystals, the HRTEM lattice image and the selected area electron diffraction (SAED) were observed on a relatively perfect Sr2IrO4 crystal (Fig. 1b). The results suggested that the Sr2IrO4 particle tends to stack the facet along [001] axis, thus exhibiting a possibility to expose (001) facet (SupplementaryFig. 1). The compositions of the particle were determined by the high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image and elemental mapping. The particle comprises of uniformly dispersed Sr, Ir, and O elements (Supplementary Fig. 2).

Fig. 1 | The thermal properties of Sr2IrO4.a, HRTEM lattice image for an undeveloped Sr2IrO4 single crystal with irregular shape. Inset shows the fast Fourier transform image. b, TEM image, HRTEM lattice image, and SAED pattern for a relatively perfect Sr2IrO4 single crystal to show the possible growth mechanism and exposed facet. The HRTEM lattice image and SAED pattern were collected on the yellow box area. c, Raman spectra of Sr2IrO4 under different temperatures. The mode v1 (a superposition of an A1g and a B2g) is a rotation of the IrO6 octahedra along the c-axis combined with an in-phase Sr displacement along c, v2 (A1gmode) is a rotation of the IrO6 octahedra along c, v3 (B2g mode) is an in-plane bending of the IrO6 octahedra, and v4 (A1gmode) is a stretching mode involving a modulation of the Ir-O (apical) distance. d-f, XRD peak shifts for (002), (200), and (110) facets of the Sr2IrO4 when varied temperatures from 25 to 90 oC. g, A scheme to show the transverse vibrations of bridging atoms under thermal stimulation, optic transverse mode (left) and acoustic transverse mode (right). h, Cooperative rocking or tilting motions of IrO6 octahedra in Sr2IrO4 resulting from the optic and acoustic transverse modes, leading to the expansion along a,b-axes and the contraction along c-axis.
The thermal response of Sr2IrO4 particles is first checked by temperature-dependent Raman spectra. As shown in Fig. 1c, there are four Raman peaks at 168.20, 307.31, 383.59, and 525.05 cm−1, labelled as v1 ~ v4. The Lorentzian function was used to fit the data and get the peak parameters (Supplementary Table 1). The mode v1(a superposition of an A1g and a B2g)is a rotation of the IrO6 octahedra along the c-axis combined with an in-phase Sr displacement along c, v2 (A1g mode) is a pure rotation of the IrO6 octahedra along c, v3 (B2g mode)is an in-plane bending of the IrO6 octahedra, and v4 (A1g mode) is a stretching mode involving a modulation of the Ir-O (apical) distance. Obviously, the v1, v2,and v4, vibration modes result from the transverse vibrations of the bridging oxygens among IrO6 units. With increasing temperatures, v2 for rotation of IrO6 units shifts from307.31 cm−1 under 25 oC to 314.12 cm−1under 90 oC, indicating the reduced tilting of IrO6 octahedra. The thermally increased intensity of peaks v1 and v4for the mixed motion is indicative of lattice distortions. Temperature-dependent XRD patterns indicated that, while heating, the (002) diffraction peak shifts towards higher angles, whereas the (200) and (110) diffraction peaks move towards lower angles (Fig. 1d-f), suggesting that the lattice contracts along c-axis and the latticeexpands along a,b-axes. Indeed, Rietveld refinements of XRD patterns further revealed that the Sr2IrO4 with I41/acd symmetry displays the contraction of the unit cell parameter c, from 25.74 Å under 25 oC to 25.72 Å under 90 oC, and the expansion of the unit cell parameter a, from 5.50 Å under 25 oC to 5.51 Å under 90 oC (Supplementary Fig. 3). The Sr2IrO4 is an anisotropic thermal expansion material, following the lattice anharmonic vibrations dominated low-frequency phonon mechanism.30 Under thermal stimulation, the IrO6 octahedra will undergo cooperative rocking motions resulting from the optic and acoustic transverse modes of bridging atoms (Fig. 1g). As a result, with elevating temperatures, the reduced tilting of IrO6 octahedra of Sr2IrO4will trigger obvious lattice distortion, the expansion along a,b-axes and the contraction along c-axis (Fig. 1h).
The Ir-O bond and d band in Sr2IrO4 under heating
To gain insight into thermal-induced variations in the local electronic and geometric structure in Sr2IrO4, we collected X-ray absorption near-edge structure (XANES), extended X-ray absorption fine structure (EXAFS), and X-ray photoelectron spectroscopy (XPS) valence band spectra (Fig. 2). In XANES spectra (Fig. 2a), the white line of Ir L3-edge originates from the electron transition from the occupied Ir 2p3/2orbital to the partially occupied Ir 5d orbital.The peak intensity of white line is related to the number of unoccupied 5d states. The white-line intensity increases when temperatures rise from 25 to 90 oC, which reveals the higher valence state of Ir when thermally increased the number of unoccupied 5d states. The EXAFS provides details about the distance of the back-scattering atom surrounding the central absorbing atom. The peak at 1.0 - 2.0 Å is assigned to scattering of the nearest 6-coordinated Ir-O atoms for the IrO6 units in Sr2IrO4 (Fig. 2b and Supplementary Fig. 4). Considering the thermal distortions, the IrO6 unit can be split into 4-coordinated Ir-O bond with bond length of 1.989 Å in the ab-plane (Ir-Oab-plane) and 2-coordinated Ir-O bond with bond length of 1.993 Å in the c-axis (Ir-Oc-axis) under 25 oC (Supplementary Table 2). Heating up to 90 oC, the Ir-Oab-plane and Ir-Oc-axisbond lengths are shortened to 1.938 Å and 1.937 Å, respectively. This is a solid evidence of compressive strain generation in the IrO6 octahedron as heating Sr2IrO4. The peak at 2.0 - 3.0 Å is attributed to the scattering of neighboring Ir-Ir atoms. The Ir-Ir bond is elongated from 2.730 Å under 25 oC to 2.744 Å under 90 oC. The elongated Ir-Ir bond is indicative of the reduced titling among IrO6octahedra. This means that the compressive strain generation in the IrO6 octahedron is a result of the transverse vibrations of bridging oxygens among IrO6octahedra. Wavelet transform (WT) analysis of EXAFS spectra, which provides both R- and k-space information and discriminates the back-scattering atoms, was undertaken to visualize and confirm the Ir-O bond inIrO6 units and Ir-Ir bond amongIrO6 units (Fig. 2c,d).
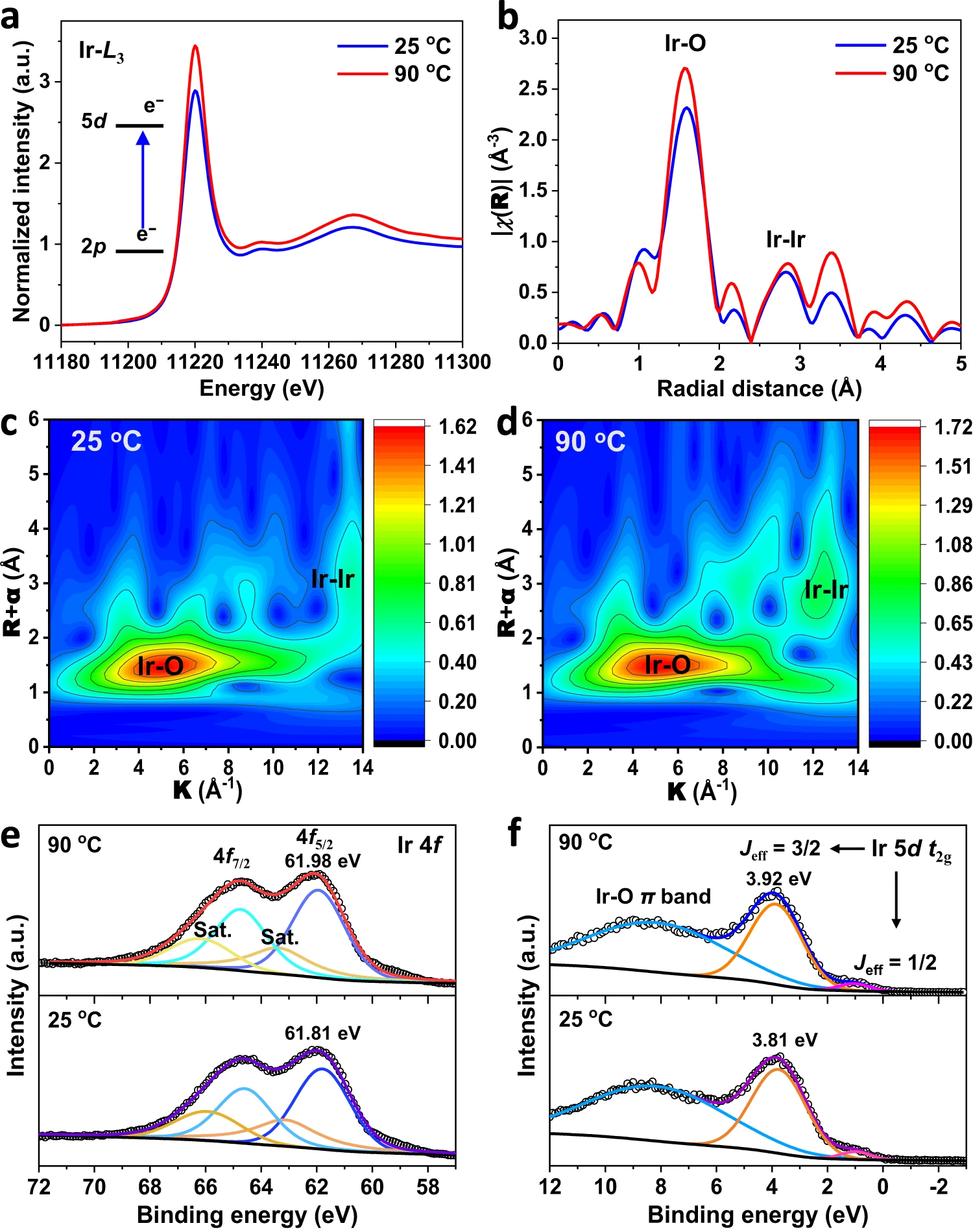
Fig. 2 |Electronic structure changes in Sr2IrO4 under thermal stimulation.a, Ir L3-edge XANES region of Sr2IrO4 measured under different temperatures. b,k3-weighted FT curves of the EXAFS data at the Ir L3-edge. c,d, Wavelet transform plots of the EXAFS data of Sr2IrO4 under 20 oC (c) and 90 oC (d). e,f, Ir 4f XPS spectra (e) and valence band spectra (f) of Sr2IrO4 under different temperatures.
The Ir 4f5/2 core-level XPS analysis indicated that the binding energy at 61.81 eV under 25 oC is assigned to the Ir4+ (Fig. 2e and Supplementary Table 3). Heating Sr2IrO4 up to 90 oC, the binding energy of Ir4+ shifted positively about 0.17 eV, indicating the increased valence state of Ir, in line with the EXAFS result. This result suggests that the compressive strain in IrO6 units is beneficial to stabilize the high valence of Ir species. Due to strong spin-orbital coupling in 5d transition-metal oxides, the low-spin Ir4+ (5d5, t2g5eg0)5d orbital in Sr2IrO4 splits the five electrons in the t2g band into four electrons occupied as the Jeff = 3/2 sub-band and to one electron occupied as the Jeff = 1/2 sub-band. The XPS valence band analysis was carried out to visualize the d-band electronic structures near the Fermi level (Ef). The valence-band XPS spectra under 25 oC show three peaks (Fig. 2f and Supplementary Table 4): the Jeff = 1/2 sub-band at 0.99 eV,the Jeff = 3/2 sub-band at 3.81 eV, and the metal-oxygen π bands at 8.17 eV. Under 90 oC, they move to 1.03, 3.92, and 8.17 eV, respectively. The increase in binding energies with increasing temperatures implies that the thermal strains in IrO6 units make the downshift of d-band away from the Ef. Indeed, the thermal-induced compressive strains shorten the Ir-O bond, thus contributing to the downshift of the d band due to the enlarged Ir-O covalency.
OER on Sr2IrO4 under heating
Next, we investigate the effects of thermal strains on OER performances. The linear sweep voltammetry (LSV) polarization curves of the Sr2IrO4 electrode were recorded under heating 1.0 M KOH electrolyte from 20 to 90 oC with inner resistance (iR) compensation of 90%, and the OER currents were normalized by the electrochemical active surface areas (ECSA) (Supplementary Fig. 5). As shown in Fig. 3a, heating from 20 to 90 oC, the potential requirement to reach 10 mA cm−2 decreased from 1.55 to 1.40 V. Considering the temperature dependence of OER thermodynamic potentials (SupplementaryTable 5),the OER overpotentials reduced from 312 mV under 20 oC to 235 mV under 90 oC. The OER performances of the Sr2IrO4 under room temperature is comparable with the known reported Ir-based catalysts (Supplementary Table 6). Notably, under heating, the OER performances of the Sr2IrO4 increase significantly. Correspondingly, the Tafel slope decreased from 90.32 mV dec−1 under 20 oC to 45.14 mV dec−1 under 90 oC (Fig. 3b). The significantly reduced Tafel slopes under higher temperatures indicate that the thermal strains, beyond the thermal-induced mass transfer, greatly accelerated OER kinetics. The positive correlation between thermal strain and OER activity was further verified by monitoring the thermal response of LSV curves (Supplementary Fig. 6). During heating and cooling electrolyte, the completely reversible LSV polarization curves witnessed that the OER performances of Sr2IrO4 are highly sensitive to the temperature.
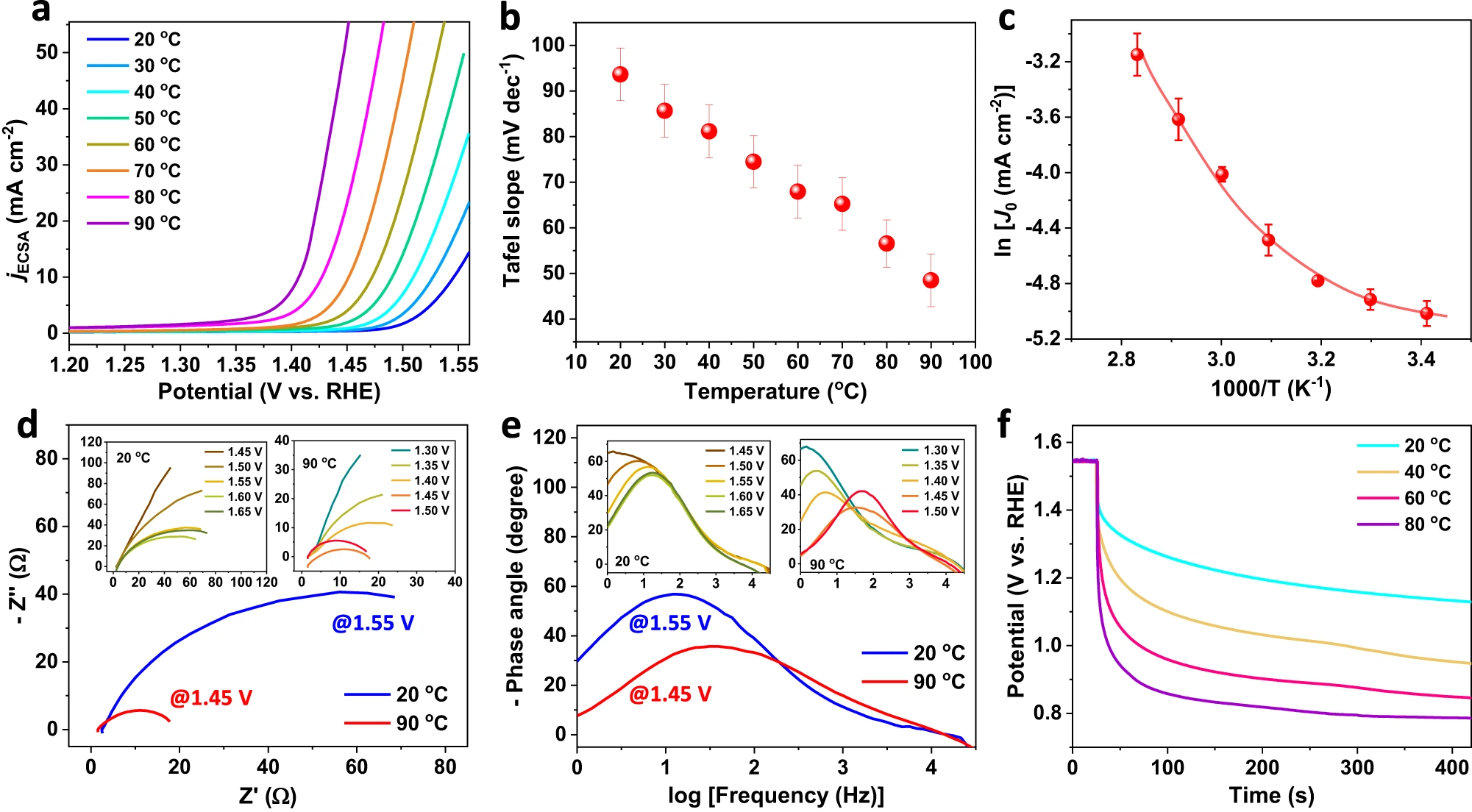
Fig. 3 | OER performances of Sr2IrO4 with thermal strains.a, LSV curvesnormalized by ECSA for Sr2IrO4 in 1.0 M KOH when varied temperatures from 20 oC to 90 oC (scan rate, 5 mV/s; mass loading, 0.4 mg cm−2; 90% iR-drop compensation is utilized; the pH of the electrolyte under different temperatures are shown in Supplementary Table 7.) b, Temperature-dependent Tafel slopes of Sr2IrO4.c, Calculated lnj0- T−1 plot with nonlinear Arrhenius relationship for OER on Sr2IrO4. d, Potential-dependent Nyquist plots at 1.55 V under 20 oC and at 1.45 V under 90 oC. Insets show the Nyquist plots when varied potentials from 1.45 to 1.65 V under 20 oC and the Nyquist plots when varied potentials from 1.3 to 1.5 V under 90 oC. e, Potential-dependent Bode plots at 1.55 V under 20 oC and at 1.45 V under 90 oC. Insets show the Bode plots when varied potentials from 1.45 to 1.65 V under 20 oC and the Bode plots when varied potentials from 1.3 to 1.5 V under 90 oC. f, OCP decay after polarization of Sr2IrO4 electrode at 1.55 V under different temperatures.
As is well-known, the thermal diffusion can promote the electrochemical OER rate through accelerating mass transfer, obeying a linear Arrhenius relationship of lnjo = −Q/(RT) + lnA (jo, exchange current density; Q, activation energy; A, pre-exponential factor; T, absolute temperature; R, universal gas constant).23 This means that the increase in OER reaction rate by thermal diffusion is strictly following a linearlnjo - T−1relationship if heating does not change the electronic states of materials. Usually, according to this linear relationship, the Q of a catalytic reaction can be calculated with a basic assumption of electronic structures of materials without dependency of temperatures. We accordingly calculated thelnjo - T−1relationship for OER of Sr2IrO4. As shown in Fig. 3c, a nonlinear lnjo- T−1relationship revealed that the Q for OER of Sr2IrO4 is a function of temperatures. This nonlinear relationship would originate from that the real-time generation of thermal strains changes the electronic states of the Sr2IrO4, thus producing a variable OER rate with the dependence of temperatures. To clearly distinguish the contributions of thermal diffusion and thermal strain during OER process, we studied heating water oxidation on SrIrO3(6H phase) with negligiblethermal strains under 20-90 oC, with just thermal diffusion to accelerate chemical reaction kinetics. Totally different from theSr2IrO4, the SrIrO3exhibits no obvious lattice distortions in XRD patterns when varying temperatures from 25 to 90 oC(Supplementary Fig. 7). This result suggests that the crystal structures of the SrIrO3 are not sensitive to the temperature. Evidently, as heating, the OER performances onSrIrO3are thermally accelerated, following a linearlnjo - T−1 Arrhenius relationship(Supplementary Figs. 8-10). The heat-induced negligible changes in binding energies for Ir, Sr, and O in the SrIrO3 confirm that heating does not affect the electronic states of the SrIrO3 (Supplementary Fig. 11). As a result,the thermally enhanced OER kinetics on the SrIrO3 electrode arise from the thermal diffusion.
The in situ electrochemical impedance spectroscopy (EIS) was performed to verify the thermally triggered strain effects on OER electron transfer. A single phase peak was detected in Bode plots and canbe assigned to the electron transfer at the interface between active center and OER intermediates (*OH,*O,*OOH)(Fig. 3d,e). Indeed, the phase angle starts at the potentials above 1.5 V under 20 oC and above 1.4 V under 90 oC, in good agreement with the OER onset potentials of LSV curves (Fig.3a). Increasing temperatures of the electrolyte, the phase peaks moved to the higher frequencies and the phase angles sharply decreased, confirming that the thermal strains accelerate the OER kinetics. The open circuit potential (Vocp) describes the quasi-Fermi level of the electrode against reference electrode when the electrode is at open-circuit state. In our case, the quasi-Fermi level of Sr2IrO4 electrode is mainly determined by the electron filling of thed band, thus providing a route to monitor the electron transfer kinetics. After polarizing the electrode at 1.55 V under different temperatures, the Vocp decay monitors the real-time evolution of quasi-Fermi level of the electrode, which is mainly dominated by the speed for electron transfer from electric double layer to d band (Fig.3f). The VOCP decay originates from the reaction between high-valence Ir species and the reducing species from electrolyte to upshift the Fermi level of the electrode and is obviously temperature-dependent. The Vocp decay stopped at a quasi-steady potential, 1.1 V for Ir4+ species under 20 oC and 0.77 V for Ird+ (d< 4) under 90 oC, as demonstrated by the XPS analysis to show low-valence Ir for electrode after OER under 90 oC (Supplementary Fig. 12).40 This fact suggests that the electron transfer and reactivity of the surface active Ir species are temperature-dependent. Higher temperatures will benefit the valence-state variation of active Ir species, indicative of the higher OER activity for Sr2IrO4 with thermal strains. Enlarging the thermal strains under higher temperatures, the rapid Vocp decay and the lower quasi-Fermi level are a result of low-barrier electron transfer.
Next, we established the plausibility of the mechanism of thermal strains promoted OER on the likely exposed facets by density functional theory (DFT) calculations. In modeling thermal-induced strains, we should ensure that the compressive strains was produced in the IrO6 units of Sr2IrO4. We firstly created the structural model of Sr2IrO4 with the thermal strains at 20 oC and 90 oC by setting the unit cell parameters with a thermal expansion coefficient of 2.8 × 10−5 K−1 for a- and b-axes and −1.2 × 10−5 K−1 for c-axis, which is obtained by fitting XRD data. However, the structural model of Sr2IrO4 createdby setting the thermal expansion coefficient is underperformance in describing the compressive strains in IrO6 units (Supplementary Fig. 13). Therefore, as a compromise, the structural model of Sr2IrO4 with I41/acd symmetrywas built by setting the unit cell parameters from the temperature-dependent XRD refinement and the atomic fractional coordinates from Ir-O bonding length in IrO6 units from EXAFS analysis at 20 and 90 oC. The lattice parameters of Sr2IrO4 are a = b = 5.50 Å, c = 25.74 Å, for Sr2IrO4 under 25 oC, and a = b = 5.51 Å, c =25.72 Å for Sr2IrO4 under 90 oC.Under 25 oC, the Ir-Oab-plane and Ir-Oc-axis bond lengths are 1.989 Å and 1.993 Å, respectively. Under 90 oC, the Ir-Oab-plane and Ir-Oc-axis bond lengths are 1.938 Å and 1.937 Å, respectively. Considering that the Sr2IrO4 particle is not a perfect crystal with nearly irregular profile, we give comprehensive consideration on selecting the likely exposed (001) and (110) facets to carry out the DFT calculations. Firstly, the (001) facet is a thermodynamically stable plane in the Sr2IrO4,41 which is able to reflect at least partially the bulk properties of Sr2IrO4. In addition, the layered Sr2IrO4 is easy to cleave along the (001) facet due to the weak interlayer interactions, increasing the probability to expose it as terminal facet. Indeed, as shown in Supplementary Fig. 1b, although the single-crystal Sr2IrO4 particles are nearly irregular, the stacking growth of (001) facets is visible as traced by dotted lines. Therefore, the DFT calculations were conducted to investigate the adsorption properties of oxygen-containing intermediates on Ir sites of Sr2IrO4(001) and (110) facets, two likely exposed facets according to the possible crystal growth mechanism and the crystallographic symmetry of Sr2IrO4.
According to the four-step OER mechanism, the Sr2IrO4 underwent the four consecutive proton-coupled electron transfer steps with *OH, *O, and *OOH intermediates. Considering the satisfied stability of Sr2IrO4 during OER (No surface reconstruction after OER i-t test at 1.45 V at 90 oC for 1 h is observed by the HRTEM in Fig. 1a) and the single Ir coordination environment on both (001) and (110) facets (Supplementary Fig. 14), we adopted the single-site adsorbate evolution mechanism (AEM) model with 100% coverage of adsorbates (100% coverage is one absorbate per one active site) on Ir active site to calculate the Gibbs free energy changes of the elementary reaction steps.In this model, the energy requirement for adsorbing the OER adsorbates onto the single active Ir site can reflect their evolution kinetics. The surface (001) and (110) slab models of Sr2IrO4 were created and stabilized by hydroxyl group passivating the unsaturated Ir sites except the Ir active site because the hydroxylation of oxides is a thermodynamically favorite process in the 1.0 M KOH electrolyte with abundant hydroxyls, thus avoiding the undesired surface reconstruction during structure optimization.In the single-site AEM model, there is no evolution of adsorbates between the adjacent sites or the simultaneous evolution of adsorbates on all Ir sites of the surface at the same time. Obviously, the single-site AEM model avoids the possible interactions between adjacent sites, thus benefiting to reflect the intrinsic catalytic activity (Supplementary Fig.15).
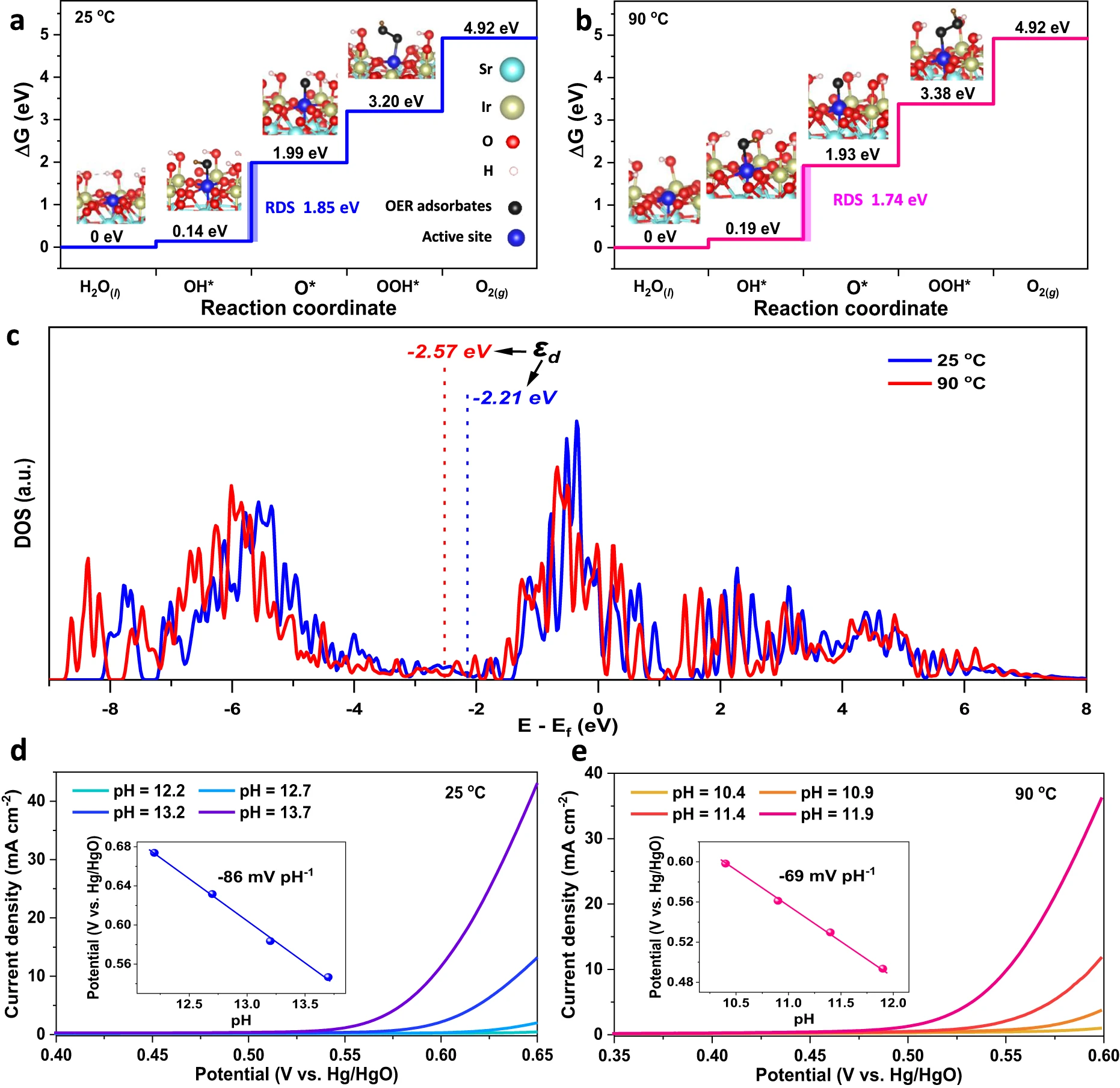
Fig. 4 | DFT calculations to show the plausibility of the thermal strains-accelerated OER kinetics.a, b, Gibbs free energy diagrams for OER intermediates (*OH, *O, *OOH) adsorbing onto the single Ir active site (the 100% coverage of OER intermediates was achieved by hydroxyl group passivating the coordinately unsaturated Ir sites except the single Ir active site) of the likely exposed Sr2IrO4(001) surface with thermal strains under 25 oC (a) and 90 oC (b), with calculated structures and rate-determining steps. Insets are the corresponding structures of the *OH, *O, and *OOH adsorptions on Ir sites. The Gibbs free energy changes were calculated without considering the effects of temperature, pH, and solvation on OER. c, The projected DOS of Ir 5d of Sr2IrO4 under 25 oC and 90 oC. The εd indicates the d-band center calculated as the first statistical moment of the d-projected DOS. d, e, The pH dependence of OER onset potentials to confirm the rate-determining step, the deprotonation of *OH on Sr2IrO4 with thermal strains under 25 oC (d) and 90 oC (e). Onset potentials were determined by the applied potentials at a constant current of 1 mA cm-2. Insets show the linear relationship of onset potential vs. pH.
We employed the PBE exchange-correlation functional with corrections of on-site Coulomb U and spin-orbit coupling (SOC) effect to describe the electronic states of Sr2IrO4 (details see the Methods). The corrections of U and SOC effect aim at compensating the underperformances in PBE describing localized 5d states. For describing the interactions between catalyst and adsorbates,Nørskov has proposed that the RPBE (a revised PBE functional for accurately describing the chemisorption energies, generating by improving the mathematical form for the exchange energy enhancement factor) is more accurate than PBE. However, the two functionals, PBE and RPBE, share the same construction logic and therefore contain the same physics and fulfill the same physical criteria, and thereforethe most limitations of PBE still remain in RPBE and the RPBE exhibits obvious improvement in describing interactions between catalyst and adsorbates. It is worth noting that the efficacy of RPBE in describing catalyst/adsorbates interactions depends on transition metals and RPBE usually reduces an overestimation ofPBE describingcatalyst/adsorbates interactions. To validate the plausibility of chemisorption energies calculated by PBE functional, we compared the energy profiles obtained with PBE + U + SOC (Fig. 4a,b) and RPBE + U + SOC (Supplementary Fig. 16) and found that they exhibit the similar efforts in describing the interactions between Sr2IrO4 and OER adsorbates, in good agreement with the previous OER calculations on IrO2, suggesting that the PBE + U + SOC approach is acceptable for describing the electronic states of Sr2IrO4 and its interactions with the OER adsorbates. The small difference in chemisorption energies calculated by PBE and RPBE probably stems from that the RPBE is mainly an advantage for the early and mid transition metals and not very much for the late transition metals.Here, it is worth noting that, our DFT calculations were based on the assumptions including the direct use of experimental crystal structure, the likely exposed (001) and (110) facets, and the single-site AEM model. In this situation, the DFT calculations just provide the information of electronic states of the Sr2IrO4 structure with strains, which do not reflect the ground-state properties of Sr2IrO4. And also the Gibbs free energy is a result of the interactions between the valence states of OER intermediates and the electronic states of the Sr2IrO4 structure with strains.
The free energy calculations on (001) facet of the Sr2IrO4 with the small thermal strains under 25 oC exhibit that the free energy difference between ΔG*OH and ΔG*O is maximum to be 1.85 eV, indicating that a minimum potential of 1.85 V has to be applied to make each step downhill in free energy. This means that the deprotonation of *OH to form *O is the rate-determining step (RDS) due to the strong adsorption of *OH (Fig. 4a). However, the potential requirement at OER RDS on the Sr2IrO4 with big thermal strains under 90 oC was significantly reduced to 1.74 V (ΔG*OH − ΔG*O = 1.74 eV) due to the weakened adsorption of *OH (Fig. 4b), resulting in a reduction in the OER overpotential. Indeed, recent theoretical studies on the OER mechanism for transition metal oxides have demonstrated that the ΔG*O− ΔG*OH difference can effectively describe their OER activities.Actually, this difference reflects the metal-oxygen binding strength.51 The optimal catalysts are required to have a moderate metal-oxygen bond strength and thus a neither too big nor too small ΔG*O− ΔG*OH difference. Our DFT calculations reveal that the thermal strains in Sr2IrO4 lead to a smaller ΔG*O− ΔG*OH, i.e., a smaller overpotential. This result suggested that the thermal strains in Sr2IrO4 can optimize the interactions between oxygen-containing OER intermediates and Ir active species.We provided the Bader charge analysis to check the interactions between Ir active site and OER intermediates. As shown in Supplementary Fig. 17,Bader charge analysis revealedthat about 0.39 - 0.54 electrons were transferred from OER intermediates into Ir active site during the adsorption to proceed, confirming that the Ir active site is able to extract electrons from OER intermediates via their interactions. The higher electron transfer numbers for *OH than *OOH under 25 - 90 oC agree well with the Sabatier principle, that is, an ideal OER site should adsorb *OOH weaker than *OH for initial reactant capture and product desorption. Indeed, considering the operation conditions (including pH, potential, temperature, and solvation effects), the Sr2IrO4 with thermal strains still exhibited the similar Gibbs energy profile with the RDS for deprotonation of *OH (Supplementary Figs. 18 and 19). The theoretical calculations exhibited the similar OER performances on both (001) and (110) facets of Sr2IrO4 with thermal strains, proving a plausible mechanism that the OER activities on Sr2IrO4 are mainly dominated by the thermally induced strain effects (Fig. 4 and Supplementary Fig. 20). These DFT results on the likely exposed facets further confirmed that the thermal strain in Sr2IrO4 is effective to accelerate OER, beyond the thermal diffusion.
The binding strengths of adsorbed oxygen-containing OER intermediates to active metal sites are closely related to the d-band center of transition metal oxides. Usually, a lower d-band center induces a weaker metal-oxygen binding. We also carried out the DFT calculations to check the changes in d-band center under thermal strains, establishing the plausibility of the mechanism as a thermally induced effect. The calculated density of states (DOS) ofSr2IrO4 exhibited that the t2g orbital is the main electronic states near the Ef (Supplementary Fig. 21). The d-band center was calculated as the first statistical moment of the d-projected DOS. The thermal strain downshifts the Ir 5d band of Sr2IrO4 away from the Ef, from −2.21 eV under 25 oC to −2.57 eV under 90 oC (Fig. 4c). The DFT results are in good agreement with the XPS valence band analysis. After the adsorption of OER intermediates onto the Ir active sites, the downshift of d band center is still dominated by the thermal strains, demonstrating that the interactions between OER intermediates and Ir active site does not change the downshift trend of d band center (Supplementary Figs.22 and 23).For OER on Sr2IrO4, the metal-oxygen bond strength depends on the interactions between the valence states of oxygen-containing OER intermediates and the Ir 5d states. After adsorption of oxygen-containing OER intermediates to Ir active species, their Ir-O orbital overlap will lead to the generation of the split Ir-O bonding (σ) and antibonding (σ*) states. Usually, the σ states, much lower than Ef, are fully filled, while the filling of the σ*states, above Ef, depends the energy level of these states relative to the Ef. The d-band center level (εd) is closely relative to the energy level of σ*states, thus affecting the electron filling of σ*states. As a consequence, the lower the εd of the Sr2IrO4 with big thermal strains, the weaker the binding of *OH to the Ir active species, because more electrons will fill the σ*states and be occupied when adsorption occurs between the Sr2IrO4 and OER intermediates.52
The thermal strains accelerated proton-coupled electron-transfer process in OER was further demonstrated by dependence of pH on the onset potentials of OER. According to the Nernst equation, for an electrochemical reaction with highly solvated protons under ambient conditions (25 oC, 1 atm), the thermodynamic potential of the electrode shifts −59 mV per pH unit as the pH is increased. In our case, in alkaline media, the fast reaction rate of 1011 mol L−1 s−1 for H3+O and OH− recombination induces that the OER kinetics is completely limited by sluggish deprotonation of *OH. As a result, the dependence of pH on the OER onset potentials is −86 mV pH−1 for Sr2IrO4 with small strains under 25 oC (Fig. 4d), significantly bigger than the theoretical value of −59 mV pH−1, confirming the frustrated deprotonation of *OH. As heating to 90 oC, the slope for pH dependence of OER onset potentials is −69 mV pH−1 (Fig. 4e), which is much close to the theoretical value of −72 mV pH−1 under 90 oC and 1 atm for completely solvated protons, indicating that the OER RDS, deprotonation of *OH to *O, on the Sr2IrO4 is effectively accelerated when heating to produce the big thermal strains under 90 oC. The thermal compressive strains in IrO6 units downshift the d-band center, which would induce that the proton in *OH tends to be solvated due to the weakened adsorption of *OH to Ir active species under larger thermal strains. Therefore, we conclude that the thermally lowered Ir 5d band center will weaken the binding strength between the OER intermediates and Ir active species and gives rise to the higher intrinsic OER activity of Sr2IrO4 under thermal stimulation. It is worth pointing out that adjusting electronic states of Sr2IrO4 by heating is different from the previous strategies, such as electronic modifications by doping, defects, or distortions. In this case, the thermal field is utilized to tune the electronic states of Sr2IrO4 via thermal strain effect. Our results imply that it is possible to apply the materials with a positive catalytic contribution of thermal strains to OER activity for creating the efficient water splitting with the simultaneous input of heat and electricity.
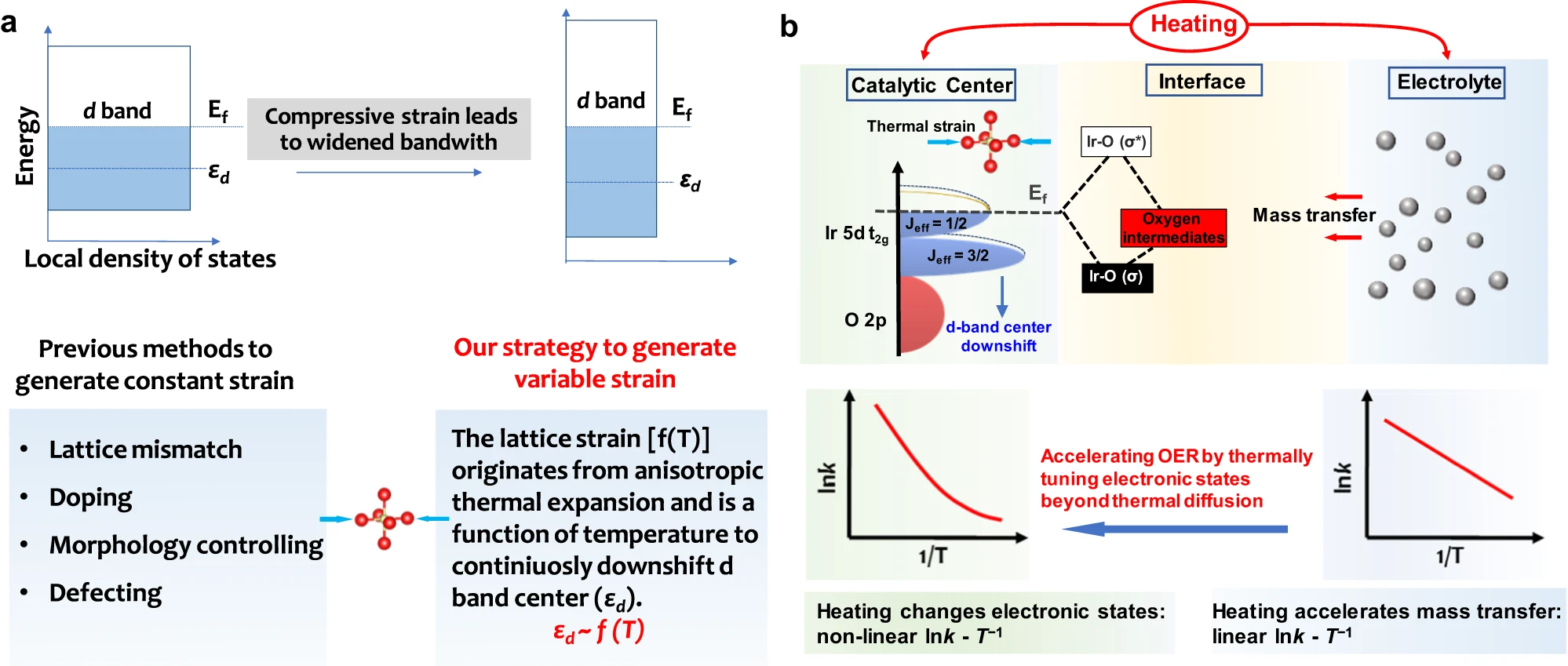
Fig. 5 | Energetics at catalyst-electrolyte interface with thermal strain effect. a, A scheme to describe the effect of compressive strain on the d band of transition metals and our thermal stimulation strategy to generate variable lattice strain. b, Effects of heating on the OER. The improved OER performances originate from that heating accelerates mass transfer and the thermal strains in IrO6 units downshift the d-band center.The strong spin-orbital coupling in the low-spin Ir4+ (5d5, t2g5eg0) 5d orbital in Sr2IrO4 splits the five electrons in the t2g band into four electrons occupied as the Jeff = 3/2 sub-band and to one electron occupied as the Jeff = 1/2 sub-band. The thermal strains downshifting d-band center is real-timely sensitive to the temperature. The thermal diffusion accelerating OER follows a linear lnk - T−1 Arrhenius relationship and the thermal strain accelerating OER follows the nonlinear lnk - T−1 Arrhenius relationship due to that downshifting d-band center will adjust the Ir-O bonding (σ) and antibonding (σ*) states to meet the Sabatier principle.
Discussion
In summary, we successfully demonstrate that the d-band center of Sr2IrO4 can be easily tuned by heating to trigger compressive strains in its IrO6 units, thus optimizing the OER kinetics by adjusting the binding strength between OER intermediates and Ir active species. Completely different from the previous routes to produce the constant strains in crystals (Fig. 5a), the thermally induced strains are easily tuned by varying temperature. As an example, the thermal compressive strains in the Sr2IrO4 crystal with anisotropic thermal expansion will effectively regulate the electronic energetics at electrode-electrolyte interface (Fig. 5b), downshifting the d-band center to accelerate the OER kinetics and breaking the linear Arrhenius relationship to achieve higher energy conversion efficiency. Our findingsindicated that heating is a possible route to tune the electronic states of materials, which may exhibit a positive catalytic contribution to OER kinetics, beyond the traditional thermal diffusion effect.
Acknowledgments
This work is supported primarily by the Scientific and Technological Innovation Project of Carbon Emission Peak and Carbon Neutrality of Jiangsu Province (No.BE2022028-1), the National Natural Science Foundation of China (Grant Nos 52272217, 22372078, 51872135, 51572121, and 21633004).
Author contributions
Y.D. and F.X. contribute equally to this work.S.Y. and Y.D. conceivedthe original concept, designed the experiments and wrote the manuscript.Y.D. and F.X. performed most characterizations and analysis. R.L. finished the temperature-dependent X-ray diffraction characterization and analysis. M.L., W.L., and Y.Y. discussed characterizations and electrochemical testing results. Z.Z. discussed the experimental ideas. All authors discussed the results.
Keywords:Heat-electricity coupling;Thermal strains;d band center;Renewable power driven hydrogen production
Website link:https://doi.org/10.1038/s41467-024-46216-9
